Table of Contents
- What is immunological evaluation of medical devices?
- The standard stack: ISO 10993-10, ISO 10993-20, GB/T 16886.10/.20, FDA, EU MDR
- Sensitisation testing: GPMT, LLNA, and the in vitro alternatives
- Irritation testing: the in vivo rabbit and the in vitro RhE replacement
- ISO 10993-20: the immunotoxicology framework
- The 28-day immunophenotyping study: the functional immune assessment
- Systemic toxicity: acute, subacute, subchronic, and the immune-system endpoints
- The FDA vs EU regulatory divergence on immunological endpoints
- BEP, BER, and the risk-based evaluation framework
- FAQ
- Our immunological evaluation capabilities
What is immunological evaluation of medical devices?
Immunological evaluation is the assessment of the potential adverse effects of a medical device or its materials on the immune system of the patient or user — the sensitisation (allergic contact dermatitis), the irritation (inflammatory response), and the systemic immunotoxicity (immune suppression, immune stimulation, or immune dysregulation). It is governed by two specific parts of the ISO 10993 / GB/T 16886 series: ISO 10993-10 Tests for irritation and skin sensitisation (the "Big Two" of the biocompatibility matrix — the two endpoints that almost every device must test) and ISO 10993-20 Immunotoxicology testing of medical devices (the framework for the functional immune assessment, triggered for devices with prolonged or permanent contact with tissue or blood, or with materials known to interact with the immune system). The output of an immunological evaluation is a dossier covering the sensitisation test (GPMT or LLNA, or the in vitro DPRA/KeratinoSens/h-CLAT alternatives), the irritation test (in vivo rabbit intracutaneous or in vitro RhE), and — when triggered — the immunotoxicology study (28-day rodent implantation with peripheral-blood and spleen immunophenotyping).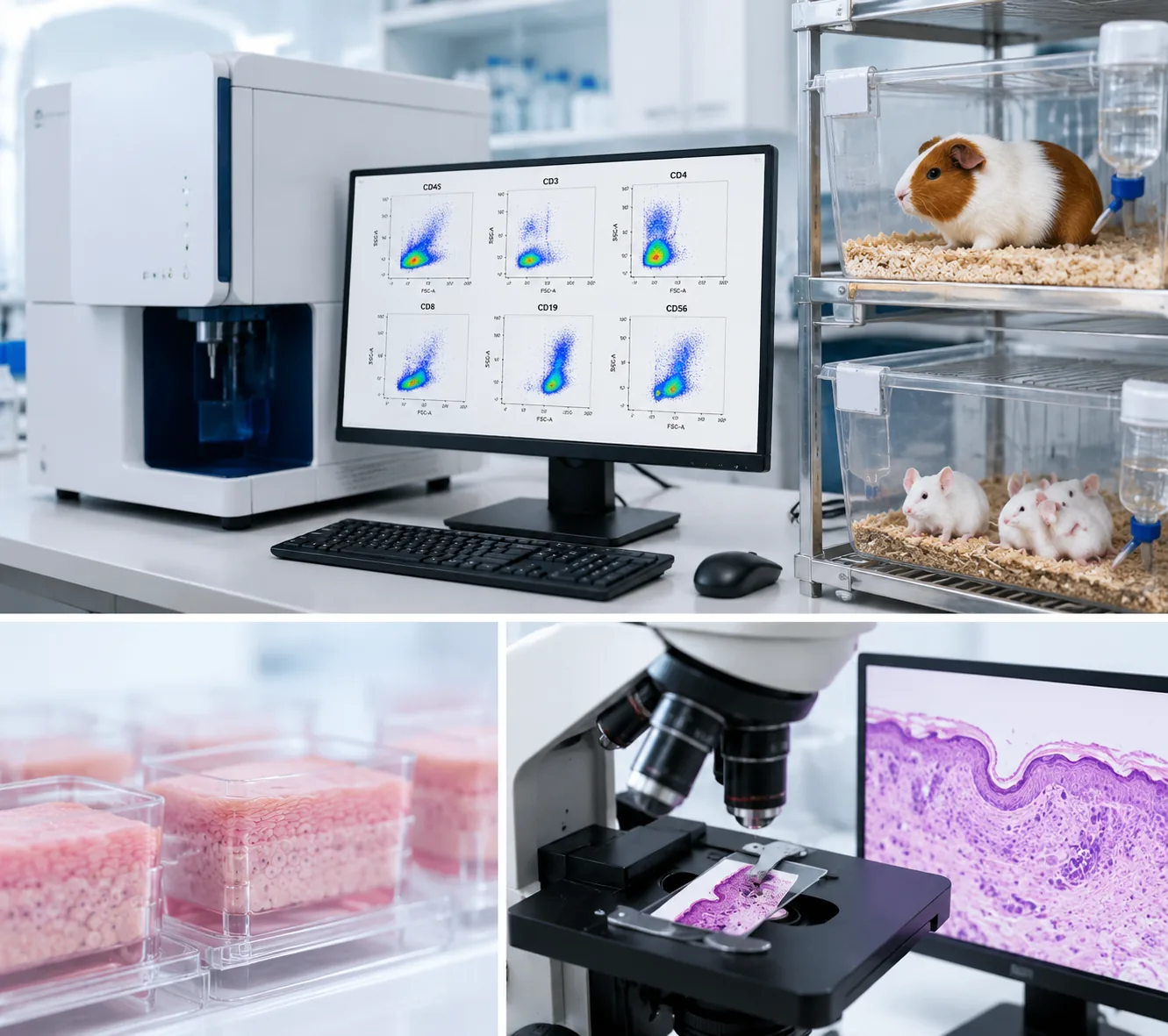
The immune system is the body's primary defence against foreign materials. A medical device introduced into the body — by surface contact (a wound dressing, an electrode), by external communication (an IV catheter, a dialyser), or by implantation (a hip prosthesis, a cardiac stent) — is recognised by the immune system as foreign, and the system responds. The response is usually benign (the immune system tolerates the device), but in some cases the device materials — leachable chemicals, residual monomers, degradation products, surface coatings, or the device's physical surface itself — can trigger an adverse immune response: a contact dermatitis (from a surface-contact device), a delayed-type hypersensitivity (from an implanted device), a systemic immune suppression (from an immunosuppressive leachable), or a systemic immune stimulation (from an adjuvant-like leachable). The immunological evaluation verifies that the device's materials do not produce any of these adverse effects at the clinical dose and duration.
The standards governing immunological evaluation span the ISO 10993-10:2010 (the international standard for irritation and sensitisation testing, with its 2024 revision aligning with the in vitro OECD TG methods), the ISO/TS 10993-20:2006 (the international technical specification for immunotoxicology testing), the GB/T 16886.10-2017 (the Chinese adoption of ISO 10993-10), the GB/T 16886.20-2017 (the Chinese adoption of ISO/TS 10993-20), the FDA Guidance on ISO 10993-1 (which still requires the in vivo rabbit intracutaneous test for irritation, not accepting the in vitro ISO 10993-23 RhE), the EU MDR 2017/745 (which accepts the in vitro RhE and the OECD TG in vitro methods), and the OECD Test Guidelines for the in vitro alternatives (TG 442C DPRA, TG 442D KeratinoSens, TG 442E h-CLAT). See our dedicated biocompatibility testing article for the broader ISO 10993 framework.
The standard stack: ISO 10993-10, ISO 10993-20, GB/T 16886.10/.20, FDA, EU MDR
A complete immunological evaluation project draws on a stack of international, Chinese, US, and EU standards and guidelines.
| Family | Standard | Scope |
|---|---|---|
| ISO 10993-10:2010 | Tests for irritation and skin sensitisation | The international standard for the Big Two immunological endpoints — irritation (intracutaneous rabbit, or RhE in vitro per ISO 10993-23) and sensitisation (GPMT, LLNA, or the in vitro OECD TG methods) |
| ISO 10993-23:2021 | Tests for irritation | The in vitro RhE (reconstructed human epidermis) standard — the replacement for the in vivo rabbit intracutaneous test |
| ISO/TS 10993-20:2006 | Immunotoxicology testing of medical devices | The framework for the functional immune assessment — when to trigger, what to test, how to interpret |
| GB/T 16886.10-2017 | 《医疗器械生物学评价 第 10 部分:刺激与皮肤致敏试验》 | The Chinese adoption of ISO 10993-10; the NMPA-mandated standard for the Big Two |
| GB/T 16886.20-2017 | 《医疗器械生物学评价 第 20 部分:医疗器械免疫毒理学试验原则和方法》 | The Chinese adoption of ISO/TS 10993-20; the framework for the functional immune assessment in China |
| FDA Guidance on ISO 10993-1 (2023) | Use of International Standard ISO 10993-1 | The FDA's biological evaluation guidance; still requires the in vivo rabbit intracutaneous test for irritation; does not fully recognise the in vitro ISO 10993-23 |
| EU MDR 2017/745 | Medical Device Regulation | The EU regulatory framework; accepts the in vitro methods (ISO 10993-23 RhE, OECD TG 442C/D/E) for the sensitisation and irritation assessment |
| OECD TG 429 / 442A | Skin Sensitisation: Local Lymph Node Assay (LLNA) | The in vivo murine sensitisation method |
| OECD TG 406 | Skin Sensitisation (Guinea Pig Maximisation Test, GPMT) | The in vivo guinea-pig sensitisation method |
| OECD TG 442C | DPRA (Direct Peptide Reactivity Assay) | The in chemico sensitisation method (the first key event of the skin-sensitisation adverse outcome pathway) |
| OECD TG 442D | KeratinoSens / LuSens | The in vitro keratinocyte activation method (the second key event) |
| OECD TG 442E | h-CLAT / U937 / MUSST | The in vitro dendritic-cell activation method (the third key event) |
| ICH S8 | Immunotoxicology Studies for Human Pharmaceuticals | The pharmaceutical immunotoxicology framework (cross-reference for the medical-device immunotoxicology) |
The single most consequential fact for a Chinese manufacturer is that GB/T 16886.10-2017 and GB/T 16886.20-2017 are the NMPA-mandated standards for the immunological evaluation. The sensitisation test (GPMT or LLNA) and the irritation test (intracutaneous rabbit or RhE) are required on almost every device with body contact per the ISO 10993-1 Annex A matrix; the immunotoxicology study (ISO 10993-20 / GB/T 16886.20) is triggered for the prolonged and permanent contact devices, the blood-contacting devices, and the devices with materials known to interact with the immune system (e.g. drug-device combination products, degradable implants).
Sensitisation testing: GPMT, LLNA, and the in vitro alternatives
The sensitisation test (the assessment of the potential to cause allergic contact dermatitis or delayed-type hypersensitivity) is the most universally required immunological endpoint. The three methods and their acceptance are summarised:
| Method | Standard | Principle | Regulatory acceptance |
|---|---|---|---|
| Guinea Pig Maximisation Test (GPMT) | ISO 10993-10 / OECD TG 406 | Intradermal induction + topical challenge in guinea pigs; the response (erythema, oedema) is scored; a positive response in the test group vs. the control indicates sensitisation | Accepted by FDA (preferred for deep-tissue devices), EU, NMPA, JP — the historical gold standard |
| Local Lymph Node Assay (LLNA) | ISO 10993-10 / OECD TG 429 / 442A | The test article is applied topically to the ears of mice; the proliferation of the draining lymph-node cells (measured by ³H-thymidine or BrdU incorporation) indicates sensitisation; the stimulation index (SI) ≥ 3 is the positive threshold | Accepted by FDA, EU, NMPA, JP — the ISO 10993-10:2010 recommended method; the preferred method for most surface-contact devices |
| DPRA (Direct Peptide Reactivity Assay) | OECD TG 442C | The test article is incubated with synthetic peptides (cysteine, lysine); the peptide depletion by the reactive chemical is measured by HPLC; a > 6.4 % mean depletion is positive | Accepted by EU (in the weight-of-evidence approach with KeratinoSens + h-CLAT); not accepted by FDA as a standalone replacement |
| KeratinoSens / LuSens | OECD TG 442D | The test article is applied to a keratinocyte cell line with a luciferase reporter gene for the Nrf2 pathway; the luciferase induction indicates keratinocyte activation (the second key event of sensitisation) | Accepted by EU (weight-of-evidence); not accepted by FDA |
| h-CLAT / U937 / MUSST | OECD TG 442E | The test article is applied to a dendritic-cell-like cell line; the upregulation of the surface markers CD86 and CD54 indicates dendritic-cell activation (the third key event) | Accepted by EU (weight-of-evidence); not accepted by FDA |
The regulatory divergence between the EU (accepting the in vitro OECD TG methods in a weight-of-evidence approach) and the FDA (still requiring the in vivo GPMT or LLNA) is the single most important practical fact for a manufacturer targeting both markets. A device heading for both markets must often run the in vivo method for the FDA and may additionally run the in vitro for the EU (to reduce the animal testing per the EU 3Rs policy).
Irritation testing: the in vivo rabbit and the in vitro RhE replacement
The irritation test (the assessment of the potential to cause an inflammatory response at the contact site) is the second universally required immunological endpoint.
| Method | Standard | Principle | Regulatory acceptance |
|---|---|---|---|
| Intracutaneous reactivity test (rabbit) | ISO 10993-10 / USP <88> | The device extract is injected intradermally at multiple sites on the rabbit back; the erythema and oedema are scored at 24, 48, 72 h; the difference from the control is the reactivity score | Accepted by FDA (the required method for FDA submissions), EU, NMPA — the historical gold standard |
| In vitro RhE (Reconstructed Human Epidermis) | ISO 10993-23:2021 | The device extract is applied to a 3D reconstructed human epidermis model (EpiDerm, SkinEthic RHE); the cell viability (MTT) after exposure is the readout; viability ≥ 50 % of the negative control is non-irritating | Accepted by EU and NMPA (for most device categories); not accepted by FDA — the FDA still requires the in vivo rabbit intracutaneous test |
The ISO 10993-23:2021 RhE method was validated by a global round-robin study (De Jong et al., Toxicology In Vitro 2018) that demonstrated the RhE models predict medical-device extract irritation with sensitivity and specificity matching or exceeding the rabbit test. The 2021 publication of ISO 10993-23 as a full International Standard (not a Technical Specification) marked the regulatory acceptance of the RhE in the EU; the FDA's non-recognition remains the practical barrier for the US-bound devices.
ISO 10993-20: the immunotoxicology framework
ISO/TS 10993-20:2006 Immunotoxicology testing of medical devices defines the framework for the functional immune assessment — the evaluation of whether the device's materials suppress, stimulate, or dysregulate the immune system beyond the sensitisation and irritation endpoints.
When is immunotoxicology triggered?
The standard specifies that immunotoxicology testing should be considered when one or more of the following conditions apply:
- The device has a prolonged or permanent contact with the body (> 30 days cumulative)
- The device is a blood-contacting device (the immune system is directly exposed to the device materials)
- The device is a degradable or resorbable implant (the degradation products may interact with the immune system)
- The device incorporates a pharmacologically active substance (drug-device combination product)
- The device incorporates a biological component (tissue-engineered product, cell therapy, biological coating)
- The device material is known to have immunological activity (e.g. latex, silicone, metal ions from implants)
- The toxicological risk assessment of the chemical characterisation (ISO 10993-18) identifies an immunologically active leachable above the threshold of toxicological concern
The framework does not mandate a single test battery; instead, it specifies a hierarchical approach: starting from the structure-activity relationship (SAR) of the materials, through the in vitro screening, to the in vivo functional tests, with each step triggered by the results of the previous.
The 28-day immunophenotyping study: the functional immune assessment
The 28-day rodent immunophenotyping study is the most commonly performed functional immune test for medical devices that trigger the immunotoxicology framework.
Study design (per ISO 10993-20 and the Mora-Leiva 2024 reference for breast implants):
- Animal model — Rat (typically Sprague-Dawley or Wistar) or mouse
- Exposure — The device extract is administered by the relevant route (IV for blood-contacting, SC/IP for tissue-contacting, or the device itself is implanted)
- Duration — 28 days (the subacute period)
- Endpoints:
- Clinical observations — daily, for signs of immune suppression (infection susceptibility, weight loss) or immune stimulation (lymphadenopathy, splenomegaly)
- Haematology — complete blood count with differential (the WBC, the lymphocyte count, the neutrophil count, the monocyte count)
- Peripheral-blood immunophenotyping (flow cytometry) — the T-cell subsets (CD3+, CD4+, CD8+), the B cells (CD45RA+), the NK cells (CD161a+); the CD4/CD8 ratio; the absolute and relative counts
- Spleen immunophenotyping — at necropsy, the spleen is harvested and the splenocyte immunophenotype is measured (same markers as the peripheral blood)
- Histopathology — the spleen, the lymph nodes (draining and distant), the thymus, the Peyer's patches, the bone marrow — examined for lymphoid depletion, lymphoid hyperplasia, or other immunopathological changes
- Organ weights — the spleen, the thymus, the lymph nodes (the immune-organ weights, compared to the control)
Acceptance — A device whose 28-day immunophenotyping shows no significant difference from the control in the immune-organ weights, the histopathology, the haematology, or the peripheral-blood and splenic immunophenotype is judged non-immunotoxic. A significant deviation triggers a follow-up functional assay (e.g. the T-cell-dependent antibody response, the NK-cell cytotoxicity assay, the delayed-type hypersensitivity challenge).
The published Mora-Leiva et al. study (Biomedical Systems, 2024/2026) — the first public ISO 10993 dataset for a breast implant — is the reference for this study design, demonstrating the 28-day immunophenotyping with CD3/CD4/CD8/CD45RA/CD161a panels in the rat. See our silicone implant testing and biocompatibility testing articles for the broader context.
Systemic toxicity: acute, subacute, subchronic, and the immune-system endpoints
The systemic toxicity studies (ISO 10993-11) include immune-system endpoints that overlap with the immunotoxicology framework:
| Study | Duration | Immune-system endpoints |
|---|---|---|
| Acute systemic toxicity | 24 h post single dose | Clinical observations; haematology; no immune-specific endpoints |
| Subacute toxicity (14-day or 28-day) | 14-28 days | Haematology (WBC, lymphocyte differential); spleen and lymph-node weights; histopathology of the lymphoid organs |
| Subchronic toxicity (90-day) | 90 days | Full haematology; full lymphoid-organ histopathology; immunophenotyping if triggered |
| Chronic toxicity (6-12 months) | 6-12 months | Full haematology; full lymphoid-organ histopathology; immunophenotyping |
The systemic toxicity studies and the immunotoxicology study are often run as a combined protocol (the 28-day study with the immunophenotyping endpoints added to the standard ISO 10993-11 28-day toxicity battery), reducing the animal use per the 3Rs principle.
The FDA vs EU regulatory divergence on immunological endpoints
| Endpoint | FDA (US) | EU (MDR) |
|---|---|---|
| Sensitisation | GPMT or LLNA (in vivo); the in vitro OECD TG 442C/D/E are not accepted as standalone replacements | In vitro OECD TG 442C/D/E accepted in a weight-of-evidence approach (the three key events of the sensitisation AOP); GPMT or LLNA as confirmatory if needed |
| Irritation | In vivo rabbit intracutaneous (the required method for FDA submissions) | In vitro RhE (ISO 10993-23:2021) accepted (and increasingly expected) for most device categories |
| Immunotoxicology | Triggered per the FDA guidance; the FDA's expectations are generally more conservative than the EU's | Triggered per ISO 10993-20; the in vitro and the in vivo methods both accepted |
A device targeting both markets must plan for both pathways — typically running the in vivo GPMT/LLNA + rabbit intracutaneous for the FDA and the in vitro OECD TG + RhE for the EU, or running the in vivo as the reference and the in vitro as the supporting data for the EU 3Rs policy.
BEP, BER, and the risk-based evaluation framework
The immunological evaluation is not a set of tests — it is a risk-based written evaluation documented in the Biological Evaluation Plan (BEP) and the Biological Evaluation Report (BER):
-
BEP (Biological Evaluation Plan) — documents the device description, the materials, the manufacturing process, the body-contact category and duration per ISO 10993-1 Annex A, the resulting list of biological endpoints to be evaluated (sensitisation, irritation, systemic toxicity, immunotoxicology), the rationale for testing or waiving each endpoint, the test methods selected, and the acceptance criteria. For the immunological endpoints, the BEP documents whether the sensitisation and irritation testing is triggered and whether the immunotoxicology (ISO 10993-20) framework is triggered.
-
BER (Biological Evaluation Report) — documents the evaluation strategy, the test results, the interpretation, and the overall conclusion. For the immunological endpoints, the BER reports the GPMT/LLNA result, the RhE/in vivo rabbit result, and (if triggered) the 28-day immunophenotyping result, with the judgement of whether the device is non-sensitising, non-irritating, and non-immunotoxic.
See our biocompatibility testing article for the broader BEP/BER framework.
FAQ
What is the difference between immunological evaluation and biocompatibility testing?
Immunological evaluation is a subset of biocompatibility testing — it covers the sensitisation, irritation, and immunotoxicology endpoints (ISO 10993-10 and ISO 10993-20). Biocompatibility testing covers all the ISO 10993-1 Annex A endpoints — cytotoxicity (ISO 10993-5), sensitisation and irritation (ISO 10993-10), systemic toxicity (ISO 10993-11), genotoxicity (ISO 10993-3), implantation (ISO 10993-6), hemocompatibility (ISO 10993-4), and chemical characterisation (ISO 10993-18). The immunological evaluation is the part that addresses the immune-system-specific effects.
What is the difference between GPMT and LLNA for sensitisation testing?
GPMT (Guinea Pig Maximisation Test) uses intradermal induction + topical challenge in guinea pigs, with erythema/oedema scoring — the historical gold standard, preferred by the FDA for deep-tissue-contact devices. LLNA (Local Lymph Node Assay) uses topical application to mouse ears, with lymph-node cell proliferation as the readout — the ISO 10993-10 recommended method, preferred for most surface-contact devices, and the more humane (fewer animals, less suffering) method per the 3Rs.
What is ISO 10993-20 and when is it triggered?
ISO/TS 10993-20:2006 Immunotoxicology testing of medical devices defines the framework for the functional immune assessment — the evaluation of whether the device suppresses, stimulates, or dysregulates the immune system. It is triggered for prolonged/permanent contact devices (> 30 days), blood-contacting devices, degradable implants, drug-device combination products, biological-component devices, and devices with immunologically active materials.
Does FDA accept the in vitro RhE test for irritation?
Not yet. The FDA still requires the in vivo rabbit intracutaneous test for FDA submissions, while the EU accepts (and increasingly expects) the in vitro RhE per ISO 10993-23:2021. A device targeting both markets must run both methods or run the in vivo as the reference.
What is the 28-day immunophenotyping study?
The 28-day rodent immunophenotyping study is the most commonly performed functional immune test. The device extract is administered for 28 days; the peripheral blood and the spleen are analysed by flow cytometry for the T-cell subsets (CD3/CD4/CD8), the B cells (CD45RA), and the NK cells (CD161a); the lymphoid organs (spleen, thymus, lymph nodes) are examined histopathologically. A device with no significant difference from the control is judged non-immunotoxic.
Our immunological evaluation capabilities
Beijing ZKGX Research (ISO/IEC 17025 accredited, CMA- and CNAS-accredited testing laboratory) provides complete immunological evaluation across the ISO 10993, GB/T 16886, FDA, and EU MDR standard stack:
- GB/T 16886.10-2017 (≡ ISO 10993-10) sensitisation and irritation — the Big Two endpoints for every device with body contact.
- GB/T 16886.20-2017 (≡ ISO/TS 10993-20) immunotoxicology — the functional immune assessment framework.
- Sensitisation — GPMT (Guinea Pig Maximisation Test, ISO 10993-10 Annex B / OECD TG 406); LLNA (Local Lymph Node Assay, ISO 10993-10 Annex C / OECD TG 429 / 442A); in vitro DPRA (OECD TG 442C), KeratinoSens (OECD TG 442D), h-CLAT (OECD TG 442E) for the EU weight-of-evidence approach.
- Irritation — in vivo rabbit intracutaneous (ISO 10993-10 / USP <88>) for the FDA; in vitro RhE (ISO 10993-23:2021, EpiDerm / SkinEthic RHE, MTT viability ≥ 50 %) for the EU; both methods run in parallel for the dual-market devices.
- Immunotoxicology (ISO 10993-20) — 28-day rodent immunophenotyping with peripheral-blood and splenic flow cytometry (CD3/CD4/CD8/CD45RA/CD161a); lymphoid-organ histopathology (spleen, thymus, lymph nodes, Peyer's patches, bone marrow); immune-organ weights; full haematology with differential.
- Systemic toxicity (ISO 10993-11) — acute (24 h), subacute (14-28 d), subchronic (90 d), chronic (6-12 months); with immune-system endpoints integrated.
- BEP and BER — written by qualified toxicologists; the immunological endpoints documented, tested, and concluded.
- FDA vs EU dual-pathway planning — the in vivo (FDA) and in vitro (EU) sensitisation and irritation methods run in parallel; the immunotoxicology framework triggered per the applicable jurisdiction.
- NMPA Class II/III registration support — GB/T 16886.10 + GB/T 16886.20 + the Chinese Pharmacopoeia requirements.
Suitable device categories include: surface-contacting devices (wound dressings, electrodes, compression bandages, surgical gloves); externally communicating devices (IV catheters, dialysers, blood oxygenators, endoscopes, guidewires); implanted devices (orthopaedic, cardiovascular, ophthalmic, dental, silicone breast implants); blood-contacting devices (stents, grafts, heart valves); drug-device combination products; tissue-engineered and biological-component devices; degradable and resorbable implants. Each project is delivered with a full data report (test protocol, instrument calibration, raw sensitisation / irritation / immunophenotyping data, histopathology, statistical analysis, classification conclusion per ISO 10993-10/-20 and GB/T 16886.10/.20) in English and/or Chinese, with CMA/CNAS stamping. Contact Beijing ZKGX Research to scope the immunological evaluation applicable to your device and target market.